Find your next collaborator at Florida International University
Welcome to FIU Discovery
FIU Discovery is Florida International University's portal for exploring and connecting with the university’s scholarly community.Use the search feature and explore the interconnected results by People, Organizations and Scholarly and Creative Works.
Click the Need Assistance button below to request assistance with establishing a collaboration.
Previous
Next

Innovation and groundbreaking discoveries are the result of decades of dedication and hard work. An opportunity to pursue a particular research topic for several years is often lifechanging. Now, five early-career FIU scientists are getting the chance with support from one of the National Science Foundation’s most competitive, prestigious programs. Faculty Early Career Development (CAREER) awards are reserved for young faculty poised to be role models in research and education. Meet these NSF CAREER awardees Trina Fletcher, Bruk Berhane, Darryl Dickerson, Ahmed Ibrahim and Amal Elawady.
Previous
Next

Stephen M. Black appointed associated dean of research at College of Medicine. Read more at FIU News
Previous
Next

FIU unveils the Cold Spray and Rapid Deposition (CoIRAD) Laboratory to help the military print replacement parts in the field. Meet Dr. Arvind Agarwal , Distinguished University Professor and Principal Investigator for the laboratory.
Previous
Next
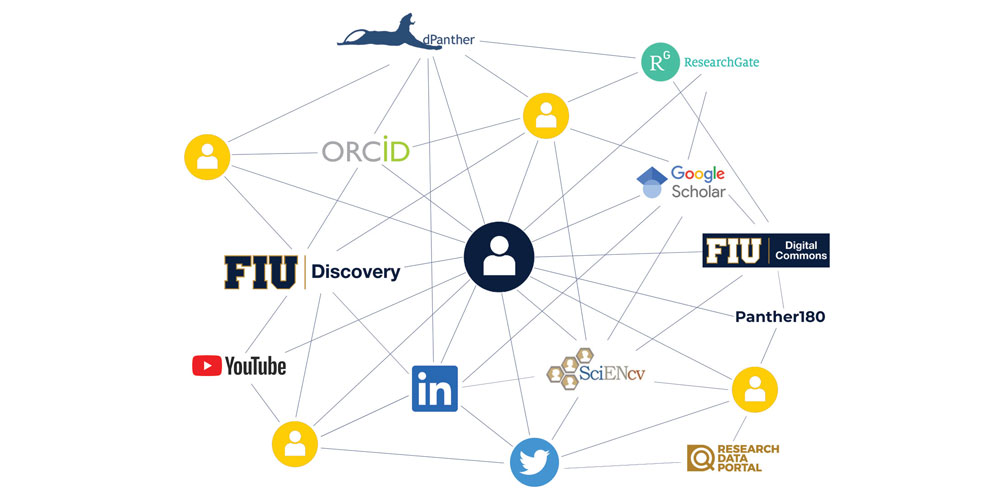
FIU Discovery can help you optimize your digital presence and reduce the amount of time spent maintaining the same scholarly productivity data across various platforms within and beyond FIU, such as ORCID, Panther180 and ScienCV. The platform is being rolled out as part of the Faculty Success Initiative, a University-wide effort that stives to promote faculty success in finding collaborators, communicating impact, and reducing burden by automating high-quality information to various digital touchpoints.
Previous
Next

Assistant professors Alexandra Coso Strong and Stephen Secules have been granted the National Science Foundation CAREER award , one of the most prestigious accolades for early-career faculty members. The two educators from the School of Universal Computing, Construction & Engineering Education (SUCCEED) will conduct research with the ultimate goal of working with engineering faculty to create positive educational experiences for students.
UN Sustainable Development Goals
The Member States of the United Nations adopted the Sustainable Development Goals by General Assembly in September 2015. The aim of these goals is to end all forms of poverty, fighting inequalities and tackling climate change while ensuring that no one is left behind. Click on a goal to the right to discover how FIU researchers and their work are contributing towards these goals.















